主要研究内容包括:
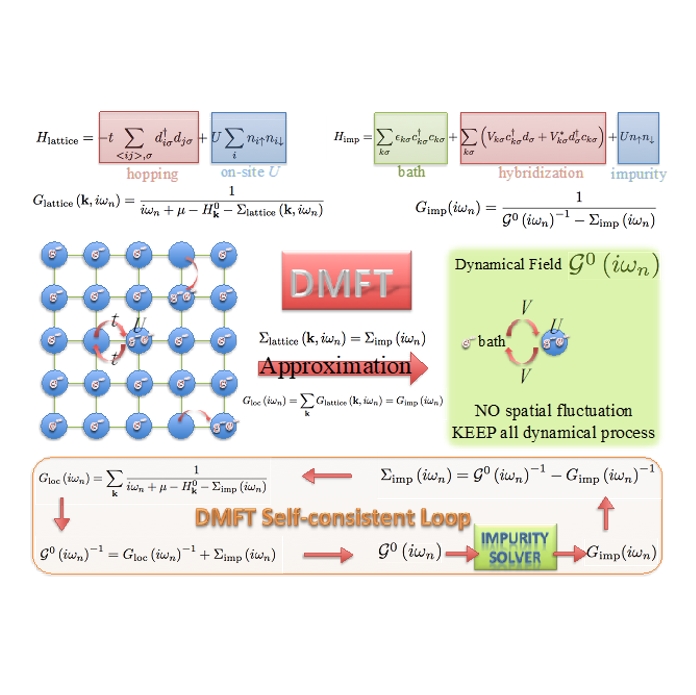
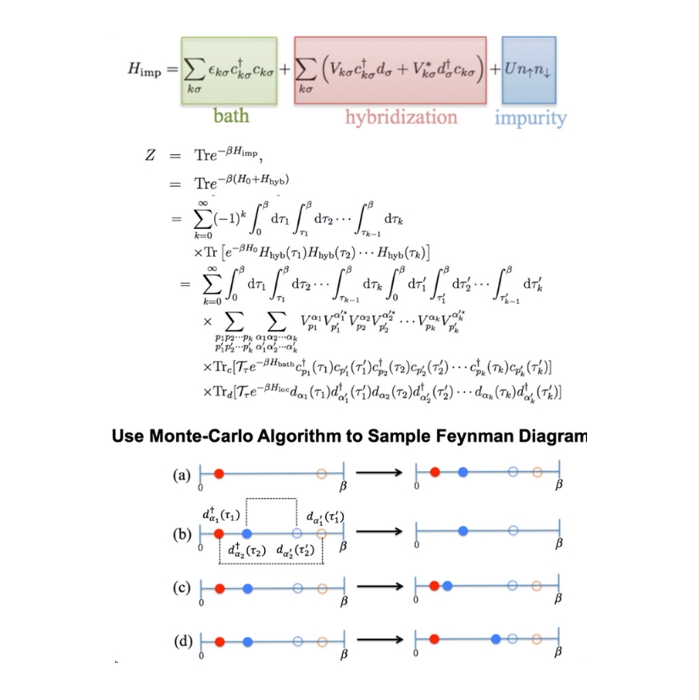
1、使用DFT+DMFT、CTQMC、DFT+Gutzwiller、DFT+Hartree-Fock等方法进行强关联材料性质的第一性原理计算;
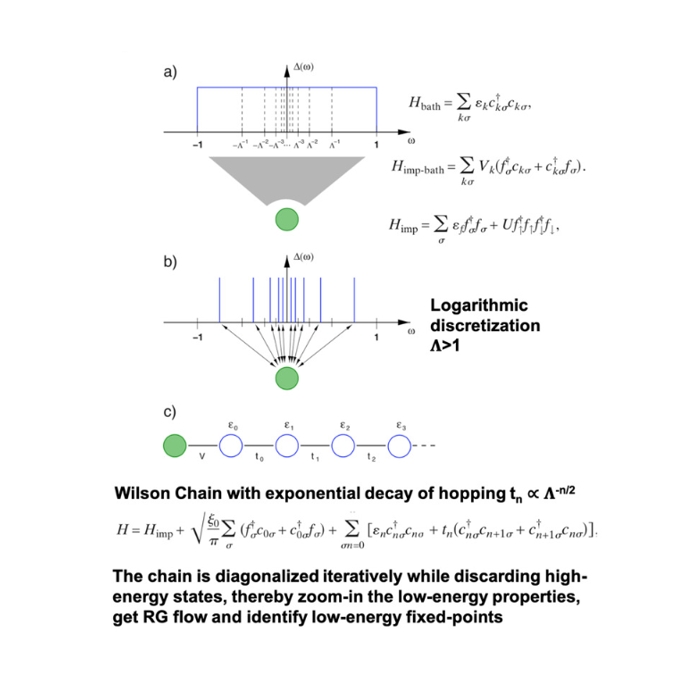
2、使用数值重整化群(NRG)、精确对角化(ED)、共形场理论(CFT)研究强关联模型;
3、x-射线谱学计算;
4、上述量子多体数值方法和软件的发展。